天使综合征是一种罕见的遗传性疾病,由母系遗传的UBE3A基因突变引起,其特征是肌肉控制能力差、言语受限、癫痫和智力障碍。尽管这种疾病目前尚无治愈方法,但北卡罗来纳大学医学院的新研究正在为治愈该疾病奠定基础。
北卡罗来纳大学医学院细胞生物学和生理学凯南杰出教授、北卡罗来纳大学神经科学中心副主任Ben Philpot 博士和他的实验室发现了一种小分子,这种分子可以安全、非侵入性地递送,并能够“开启”全脑范围内休眠的父系遗传UBE3A基因拷贝,从而产生正常的蛋白质和细胞功能,相当于一种针对天使综合征患者的基因疗法。
“我们发现的这种化合物在动物模型的发育大脑中表现出极好的吸收率,”天使综合征领域的顶尖专家菲尔波特说道。“在开始临床试验之前,我们还有很多工作要做,但这种小分子为开发一种安全有效的天使综合征治疗方法提供了一个极好的起点。”
但北卡罗来纳大学医学院细胞生物学和生理学杰出教授、神经科学中心主任马克·齐尔卡 (Mark Zylka) 表示,这些发表在《自然通讯》上的结果标志着该领域的一个重大里程碑。他补充说,目前还没有其他小分子化合物能为 Angelman 带来如此好的前景。
与囊性纤维化和镰状细胞性贫血等其他单基因疾病不同,安格曼综合征具有独特的遗传特征。研究人员发现,患有这种疾病的儿童缺少母系遗传的UBE3A基因拷贝,而父系遗传的UBE3A基因拷贝在神经元中处于休眠状态,就像在神经正常个体中一样。通常,UBE3A有助于调节重要蛋白质的水平;缺少工作拷贝会导致大脑发育严重中断。
由于尚不完全清楚的原因,UBE3A的父系基因拷贝通常在整个大脑的神经元中处于“关闭”状态。因此,当UBE3A基因的母系基因拷贝发生突变时,会导致大脑中 UBE3A 蛋白质的损失。Philpot 和其他研究人员推测,开启UBE3A的父系基因拷贝可能有助于治疗这种疾病。
这项研究的第一作者、Philpot 实验室的博士后研究员 Hanna Vihma 博士和同事从辉瑞化学遗传学库中筛选了 2,800 多种小分子,以确定是否可以在患有 Angelman 综合征的小鼠模型中有效开启父系UBE3A 。
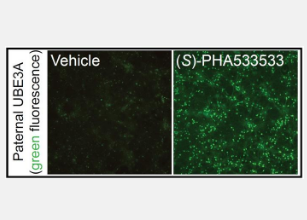
研究人员用荧光蛋白对小鼠神经细胞进行基因改造,当父系UBE3A基因被激活时,该荧光蛋白会发光。在用 2,800 多种小分子处理神经元 72 小时后,研究人员将数千个经过处理的细胞与用拓扑替康处理的细胞进行了比较。拓扑替康是一种已知的小分子,可以激活父系UBE3A,但在动物模型中缺乏治疗价值。
( S )-PHA533533 是一种之前开发为抗肿瘤药物的化合物,它使神经元发出与拓扑替康诱导的荧光相媲美的荧光,这意味着它的效果足以成功激活父系UBE3A。研究人员能够使用来自患有 Angelman 综合征的人的诱导性多能干细胞证实相同的结果,表明这种化合物具有临床潜力。
此外,研究人员还发现 ( S )-PHA533533 在发育中的大脑中具有极好的生物利用度,这意味着它可以轻松到达目标并停留在目标区域。值得注意的是,之前针对 Angelman 综合征的基因疗法的生物利用度更有限。
“我们之前曾表明,拓扑异构酶抑制剂拓扑替康在小鼠模型中的生物利用度非常低,”维玛说。“我们能够证明 ( S )-PHA533533 具有更好的吸收率,并且相同的小分子可以在人类衍生的神经细胞中转化,这是一个重大发现。这意味着它或类似的化合物具有治疗儿童的真正潜力。”
尽管 ( S )-PHA533533 显示出良好的前景,但研究人员仍在努力确定细胞内导致药物产生预期效果的精确靶点。Philpot 及其同事还需要进行进一步研究,以改进药物的药物化学,以确保该化合物(或其另一种版本)在未来临床环境中使用时安全有效。
“这不太可能是我们用于临床的确切化合物,”Philpot 说道。Philpot 实验室与 Jeff Aubé 博士实验室的药物化学家一起,正在努力寻找具有改进的药物特性和安全性的类似分子。“不过,这为我们提供了一种化合物,我们可以利用它创造出一种更好的化合物,并将其用于临床。”