分子动力学 (MD) 模拟已成为不断发展的分子生物学和药物开发领域的强大工具。虽然存在许多 MD 模拟技术,但在研究蛋白质的折叠(或“构象”)或蛋白质与配体之间的相互作用时,并行级联选择 MD (PaCS-MD) 是一种特别有用的技术。
该方法的关键在于并行运行多个 MD 模拟,从而同时探索不同的可能构象。使用精心设计的选择标准,可以在“时间快照”中自动检测有希望的构象并进行进一步研究。这种策略极大地加速了关键分子相互作用和动态过程的发现,帮助科学家了解蛋白质功能运动。
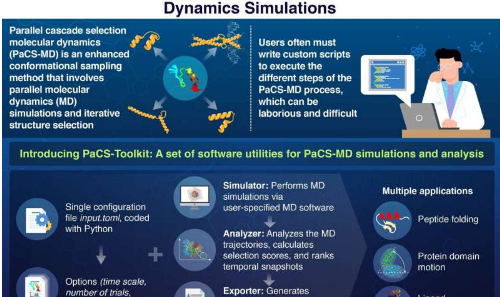
然而,PaCS-MD 的障碍之一是用户必须编写自定义脚本来执行所需的 MD 模拟。在这些脚本中,他们必须指定初始条件和目标特征,选择要使用的MD软件,实施快照排名程序,并为下一个模拟周期准备初始结构。这个过程可能非常复杂且容易出错,为有兴趣使用 PaCS-MD 的科学家设置了相当大的进入障碍。
幸运的是,日本东京工业大学生命科学技术学院的一组研究人员最近着手解决这个问题。在由 Akio Kitao 教授领导的《物理化学杂志 B》上发表的最新研究中,该团队开发了一个名为 PaCS-Toolkit 的软件包,以使 PaCS-MD 更易于使用。
PaCS-Toolkit 的一个显着优点是整个仿真过程是通过单个配置文件设置的。在此文件中,用户指定模拟的重要参数,包括 PaCS-MD 的类型、并行运行的 MD 模拟的数量以及要跟踪的蛋白质残基或原子,作为并行分支的选择标准。
PaCS-Toolkit 采用此配置文件以及标准 MD 输入文件,并根据指定的 MD 软件运行 PaCS-MD 模拟。值得注意的是,由于该软件包是用 Python(一种流行的编程语言)编写的开放软件,用户可以帮助改进 PaCS-Toolkit 并扩展其功能。
“我们的工具包保持了灵活性,因此可以通过在 Python 中引入负责的类来添加新功能、库和 MD 软件。可以使用这种语言进行编程的用户应该能够修改 PaCS-Toolkit 的代码并根据需要实现新方法, “北尾教授说。
PaCS-Toolkit 的另一个重要优势在于其对不同计算环境的优化和兼容性,无论是具有消息传递接口 (MPI) 的超级计算机阵列、配备多个显卡 (GPU) 的服务器,还是笔记本电脑等个人计算机。
“PaCS-Toolkit 大量整合了使用 MPI、GPU 和 Python 多处理包的并行化,从而能够根据可用的计算资源优化计算时间,”Kitao 教授解释道。
为了展示其工具包的潜力,研究人员针对三种不同的应用进行了 PaCS-MD 模拟。其中包括小蛋白 chignolin 的折叠、SARS-CoV-2 酶中蛋白质结构域的运动以及配体与重要腺苷受体的解离。
“总而言之,我们的结果表明 PaCS-Toolkit 可以轻松地用于模拟不同类型分子系统的各种动力学,”Kitao 教授总结道。
该工具包可以帮助释放 PaCS-MD 模拟在各个领域的真正潜力,使感兴趣的研究人员能够阐明复杂的分子过程并加速药物发现。